2.2.6. Dispersion Coefficients Near Surfaces¶
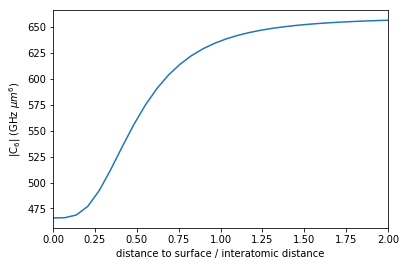
In this tutorial we reproduce the results depicted in Figure 5 from J. Block and S. Scheel “van der Waals interaction potential between Rydberg atoms near surfaces” Phys. Rev. A 96, 062509 (2017). We calculate the van der Waals \(C_6\)-coefficient between two Rubidium Rydberg atoms that are equidistantly placed in front of a perfect mirror (i.e. in horizontal alignment in front of a perfectly conducting plate). One finds that the relevant length scale is interatomic distance devided by distance from surface and that for decreasing surface distance the \(C_6\) coefficient is significantly reduced.
As described in the introduction, we start our code with some preparations and load the necessary modules.
[1]:
%matplotlib inline
# Arrays
import numpy as np
# Plotting
import matplotlib.pyplot as plt
# Operating system interfaces
import os
# pairinteraction :-)
from pairinteraction import pireal as pi
# Create cache for matrix elements
if not os.path.exists("./cache"):
os.makedirs("./cache")
cache = pi.MatrixElementCache("./cache")
The plate lies in the \(xy\)-plane with the surface at \(z = 0\). The atoms lie in the \(xz\)-plane with \(z>0\).
We can set the angle between the interatomic axis and the z-axis theta and the center of mass distance from the surface distance_surface. distance_atom defines the interatomic distances for which the pair potential is plotted. The units of the respective quantities are given as comments.
Be careful: theta = np.pi/2 corresponds to horizontal alignment of the two atoms with respect to the surface. For different angles, large interatomic distances distance_atom might lead to one of the atoms being placed inside the plate. Make sure that distance_surface is larger than distance_atom*np.cos(theta)/2
[2]:
theta = np.pi / 2 # rad
distance_atoms = 10 # µm
distance_surface = np.linspace(
distance_atoms * np.abs(np.cos(theta)) / 2, 2 * distance_atoms, 30
) # µm
Next we define the state that we are interested in using pairinteraction’s StateOne class . As shown in Figures 4 and 5 of Phys. Rev. A 96, 062509 (2017) we expect changes of about 50% for the \(C_6\) coefficient of the \(|69s_{1/2},m_j=1/2;72s_{1/2},m_j=1/2\rangle\) pair state of Rubidium, so this provides a good example.
We set up the one-atom system using restrictions of energy, main quantum number n and angular momentum l. This is done by means of the restrict... functions in SystemOne.
[3]:
state_one1 = pi.StateOne("Rb", 69, 0, 0.5, 0.5)
state_one2 = pi.StateOne("Rb", 72, 0, 0.5, 0.5)
# Set up one-atom system
system_one = pi.SystemOne(state_one1.getSpecies(), cache)
system_one.restrictEnergy(
min(state_one1.getEnergy(), state_one2.getEnergy()) - 30,
max(state_one1.getEnergy(), state_one2.getEnergy()) + 30,
)
system_one.restrictN(
min(state_one1.getN(), state_one2.getN()) - 3,
max(state_one1.getN(), state_one2.getN()) + 3,
)
system_one.restrictL(
min(state_one1.getL(), state_one2.getL()) - 1,
max(state_one1.getL(), state_one2.getL()) + 1,
)
The pair state state_two is created from the one atom states state_one1 and state_one2 using the StateTwo class.
From the previously set up system_one we define system_two using SystemTwo class. This class also contains methods set.. to set angle, distance, surface distance and to enableGreenTensor in order implement a surface.
[4]:
# Set up pair state
state_two = pi.StateTwo(state_one1, state_one2)
# Set up two-atom system
system_two = pi.SystemTwo(system_one, system_one, cache)
system_two.restrictEnergy(state_two.getEnergy() - 3, state_two.getEnergy() + 3)
system_two.setAngle(theta)
system_two.setDistance(distance_atoms)
system_two.setSurfaceDistance(distance_surface[0])
system_two.enableGreenTensor(True)
system_two.buildInteraction()
We calculate the \(C_6\) coefficients. The energyshift is given by the difference between the interaction energy at given surface_distance and the unperturbed energy of the two atom state state_two.getEnergy(). The \(C_6\) coefficient is then given by the product of energyshift and distance_atoms**6.
idx is the index of the two atom state. The command getOverlap(state_two, 0, -theta, 0) rotates the quantisation axis of state_two by theta around the y-axis. The rotation is given by the Euler angles (0, -theta, 0) in zyz convention. The negative sign of theta is needed because the Euler angles used by pairinteraction represent a rotation of the coordinate system. Thus, the quantisation axis has to be rotated by the inverse angle.
[5]:
# Calculate C6 coefficients
C6 = []
for d in distance_surface:
system_two.setSurfaceDistance(d)
system_two.diagonalize()
idx = np.argmax(system_two.getOverlap(state_two, 0, -theta, 0))
energyshift = system_two.getHamiltonian().diagonal()[idx] - state_two.getEnergy()
C6.append(energyshift * distance_atoms**6)
[6]:
# Plot results
plt.plot(distance_surface / distance_atoms, np.abs(C6))
plt.xlim(min(distance_surface / distance_atoms), max(distance_surface / distance_atoms))
plt.xlabel("distance to surface / interatomic distance")
plt.ylabel(r"|C$_6$| (GHz $\mu m^6$)")
plt.show()